Bioinformatics in CUDA
2025 | GitHubBioinformatics Implementations
A personal exploring high-performance sequence processing and GPU acceleration in C++ and CUDA.
Motivation
I got interested in bioinformatics after learning about Dor Yeshorim and genetic screening in general. After experimenting with implementing Smith-Waterman, the project turned into a parallel programming one where I extended it to implement Hamming Distance and Needleman Wunsch in CUDDA. These algorithms are pretty classic CS problems that benefit a lot from being parallelized.
A lot of the profiling and reliability techniques come from what I learned in CSE 491: Software Performance Engineering. While the class used C, tools like Valgrind and perf are directly applicable to C++ for memory and performance analysis.
The systems design was inspired partly by my AWS experience and partly by the TensorNEAT library . While cloud computing is different from pure C++ systems, many intuitions about throughput, asynchronous processing, and scalability carried over. Going through the TensorNEAT library for one of my research project taught me a lot about system design, which I tried to emulate in this project.
Implemented Algorithms
-
Hamming Distance
Computes the number of positions at which two fixed-length sequences differ. Highly parallelizable and ideal for GPU-accelerated batch processing, making it suitable for large-scale genotype or sequence comparisons. More info can be found here -
Needleman-Wunsch
Aligns two sequences over their full length using match/mismatch scoring and gap penalties. Requires more careful memory management and traceback than Hamming Distance. More info can be found here -
Smith-Waterman
Finds the best matching subsequence between two sequences. Adds complexity with zeroing negative scores and tracking local alignments efficiently on GPU. More info can be found here
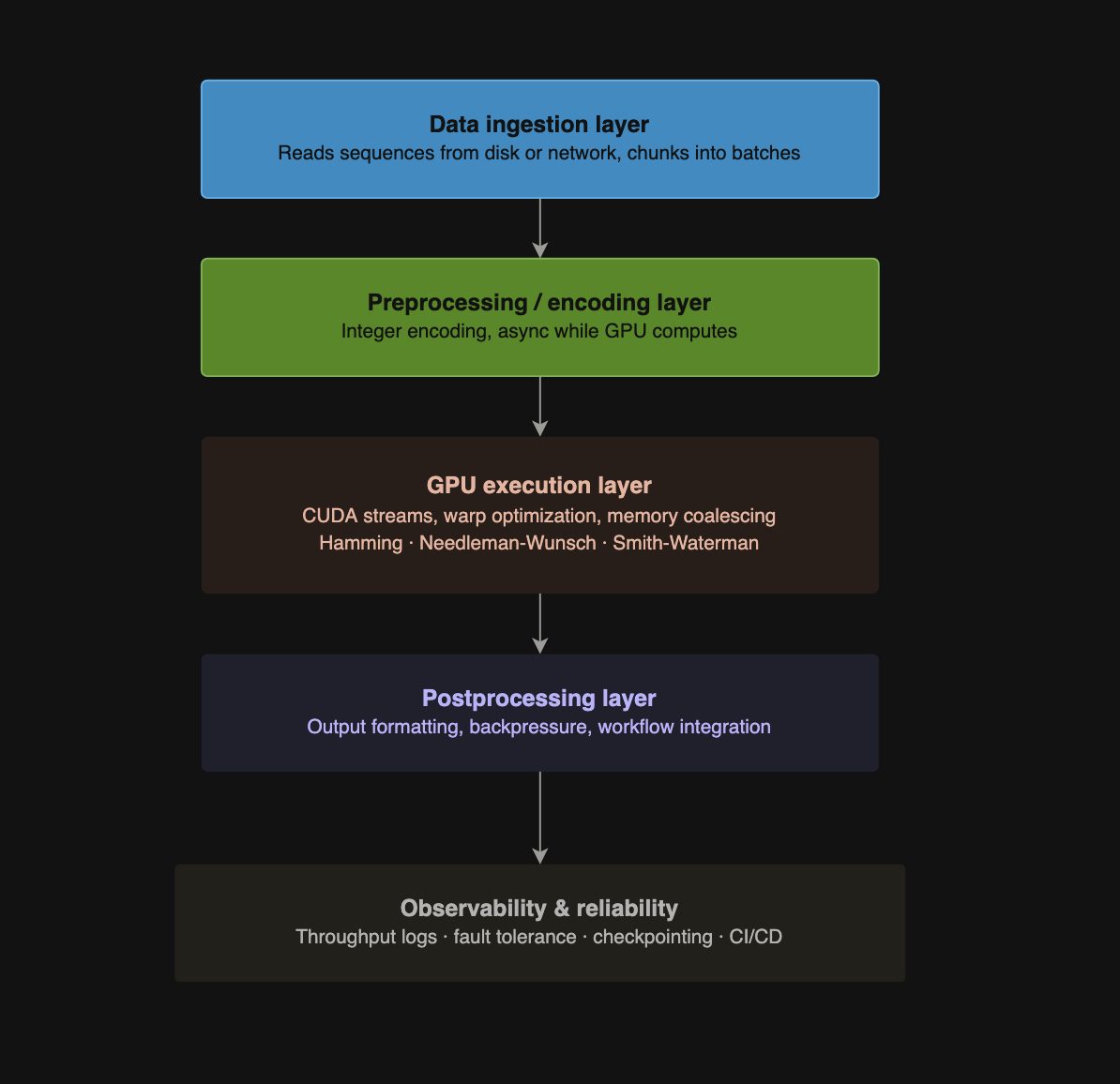
GPU-Accelerated Streaming Pipeline
Designed for high-throughput sequence processing at terabyte scale:
-
Data Ingestion Layer
- Reads sequences from disk or network.
- Chunks sequences into GPU-friendly batches for asynchronous processing.
-
Preprocessing / Encoding Layer
- Converts sequences into integer encoding suitable for GPU computation.
- Runs asynchronously while GPU is computing to avoid idle time.
-
GPU Execution Layer
- Executes alignment algorithms using multiple CUDA streams for overlapping copy and compute.
- Optimized memory access, warp utilization, and thread parallelism for maximum throughput.
-
Postprocessing Layer
- Converts GPU results back to usable formats.
- Handles output writing, backpressure, and downstream workflow integration.
-
Observability & Reliability
- Logs throughput, latency, and memory usage.
- Uses fault tolerance and checkpointing to handle long-running terabyte-scale jobs.
Datasets used: Kaggle DNA Sequences dataset and 1000 Genomes Project (50 Genomes)
The Kaggle dataset has 6 classes with a total of 4380 sequences. I structured this pipeline as a kind of classification problem where I randomly sample a genome and use Smith-Waterman or whatever algorithm to predict its class. This dataset is pretty small and thus CPU performs a lot better than CUDA.
Each indivial genome is quite large. A single genome in the 1000 Genomes Project dataset was about 3 GB. Even though I had 50 TB of strach space, I chose to keep my experiments smaller and show their scalability. Projections for each algorithm can be found in its corresponding README.
Performance & Reliability
- All profiling was done on entire algorithms. For example, for Smith Waterman, I only parallelized the DP scoring matrix part, but the runtime I recorded includes initialization and traceback.
- Achieved 30× speedup over CPU baselines on large datasets with 1B+ sequences.
For my final test, I ran the simulation on the UniProt Dataset capping it at 100,000 sequences. The dataset is a lot bigger, but for this benchmark I thought 100,000 sequences would suffice.
CPU Smith Waterman: 370.095s CUDA Smith Waterman: 9.8s
This is a 37x increase in speed. For Smith Waterman I only bencharmked the create scoring matrix function because it is the only parallelized part. The traceback wasn't included in benchmarking. However, this is a signification speed up, especially since creating the matrix the most compute intensive aprt of the algorithm.
For relibability I got 90%+ unit test coverage using GoogleTest and automated CI/CD with GitHub Actions. This includes building, formatting with clang-format, and testing.
Project Structure
/src/algorithms– Implementations of algorithms. Each algorithm contains a README with more information about the algorithm and profiling./src/pipeline– Highly modular pipeline for streaming in data. Supports GPU and CPU./utils– Utilities libraries./tests– Unit tests using GoogleTest. Each algorithm and the pipeline has its own tests.CMakeLists.txt– Modular build system with clear dependencies for extensibility.
Formatting
Trying my best to keep some sort of coherent coding style. Mainly using Meta's C++ styling. Also trying to use to use docstrings and stuff. Most of my recent classes haven't been requiring them so I've been pretty lazy about it. But since I'm about to graduate, I'm trying to get at writing more professional looking code.
Future Work
- Add more sequence algorithms (e.g., BLAST-style heuristics, Smith-Waterman variants).
- Extend pipeline for multi-GPU and multi-node scaling.
- Integrate with cloud storage for large-scale sequence datasets.